By clicking on the link, you will be leaving the official Royal Philips ("Philips") website. Any links to third-party websites that may appear on this site are provided only for your convenience and in no way represent any affiliation or endorsement of the information provided on those linked websites. Philips makes no representations or warranties of any kind with regard to any third-party websites or the information contained therein.
Dose excess and particulate downstream possibly results in a delay of wound healing, loss of microcirculation and creation of aneurysms¹⁰⁻¹³. Stellarex is the only low dose DCB with a statistically significant treatment effect in femoropopliteal lesions².
Effective low drug dose matters
Dose excess and particulate downstream possibly results in a delay of wound healing, loss of microcirculation and creation of aneurysms¹⁰⁻¹³. Stellarex is the only low dose DCB with a statistically significant treatment effect in femoropopliteal lesions².
Effective low drug dose matters
Dose excess and particulate downstream possibly results in a delay of wound healing, loss of microcirculation and creation of aneurysms¹⁰⁻¹³. Stellarex is the only low dose DCB with a statistically significant treatment effect in femoropopliteal lesions².
Dose excess and particulate downstream possibly results in a delay of wound healing, loss of microcirculation and creation of aneurysms¹⁰⁻¹³. Stellarex is the only low dose DCB with a statistically significant treatment effect in femoropopliteal lesions².
Proven performance in calcium
Proven performance in calcium
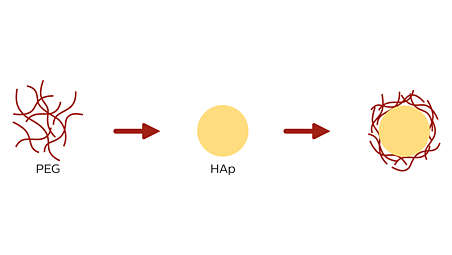
PEG forms strong ionic bonds with hydroxyl apatite (HAp), the primary component of calcified atherosclerotic lesions¹⁴. PEG’s affinity for HAp may result in limited PTX washout in the presence of calcium.
Proven performance in calcium
PEG forms strong ionic bonds with hydroxyl apatite (HAp), the primary component of calcified atherosclerotic lesions¹⁴. PEG’s affinity for HAp may result in limited PTX washout in the presence of calcium.
Proven performance in calcium
PEG forms strong ionic bonds with hydroxyl apatite (HAp), the primary component of calcified atherosclerotic lesions¹⁴. PEG’s affinity for HAp may result in limited PTX washout in the presence of calcium.
PEG forms strong ionic bonds with hydroxyl apatite (HAp), the primary component of calcified atherosclerotic lesions¹⁴. PEG’s affinity for HAp may result in limited PTX washout in the presence of calcium.
Sustained tissue Residency
Sustained tissue Residency
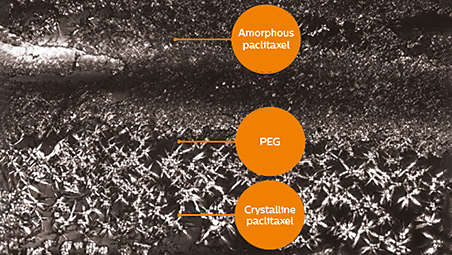
Hybrid paclitaxel offers prompt drug transfer and sustained tissue residency through 28 day restenotic window¹⁵⁻¹⁶.
Sustained tissue Residency
Hybrid paclitaxel offers prompt drug transfer and sustained tissue residency through 28 day restenotic window¹⁵⁻¹⁶.
Sustained tissue Residency
Hybrid paclitaxel offers prompt drug transfer and sustained tissue residency through 28 day restenotic window¹⁵⁻¹⁶.
Dose excess and particulate downstream possibly results in a delay of wound healing, loss of microcirculation and creation of aneurysms¹⁰⁻¹³. Stellarex is the only low dose DCB with a statistically significant treatment effect in femoropopliteal lesions².
Effective low drug dose matters
Dose excess and particulate downstream possibly results in a delay of wound healing, loss of microcirculation and creation of aneurysms¹⁰⁻¹³. Stellarex is the only low dose DCB with a statistically significant treatment effect in femoropopliteal lesions².
Effective low drug dose matters
Dose excess and particulate downstream possibly results in a delay of wound healing, loss of microcirculation and creation of aneurysms¹⁰⁻¹³. Stellarex is the only low dose DCB with a statistically significant treatment effect in femoropopliteal lesions².
Dose excess and particulate downstream possibly results in a delay of wound healing, loss of microcirculation and creation of aneurysms¹⁰⁻¹³. Stellarex is the only low dose DCB with a statistically significant treatment effect in femoropopliteal lesions².
Proven performance in calcium
Proven performance in calcium
PEG forms strong ionic bonds with hydroxyl apatite (HAp), the primary component of calcified atherosclerotic lesions¹⁴. PEG’s affinity for HAp may result in limited PTX washout in the presence of calcium.
Proven performance in calcium
PEG forms strong ionic bonds with hydroxyl apatite (HAp), the primary component of calcified atherosclerotic lesions¹⁴. PEG’s affinity for HAp may result in limited PTX washout in the presence of calcium.
Proven performance in calcium
PEG forms strong ionic bonds with hydroxyl apatite (HAp), the primary component of calcified atherosclerotic lesions¹⁴. PEG’s affinity for HAp may result in limited PTX washout in the presence of calcium.
PEG forms strong ionic bonds with hydroxyl apatite (HAp), the primary component of calcified atherosclerotic lesions¹⁴. PEG’s affinity for HAp may result in limited PTX washout in the presence of calcium.
Sustained tissue Residency
Sustained tissue Residency
Hybrid paclitaxel offers prompt drug transfer and sustained tissue residency through 28 day restenotic window¹⁵⁻¹⁶.
Sustained tissue Residency
Hybrid paclitaxel offers prompt drug transfer and sustained tissue residency through 28 day restenotic window¹⁵⁻¹⁶.
Sustained tissue Residency
Hybrid paclitaxel offers prompt drug transfer and sustained tissue residency through 28 day restenotic window¹⁵⁻¹⁶.
1. Lyden S, ILLUMENATE trial update, oral presentation, AMP Aug 2019.. Granada JF. Future directions, clinical applications and local drug delivery technologies. Presented at the Transcatheter Cardiovascular Therapeutics (TCT) 25th Annual Scientific Symposium; October 27-November 1, 2013; San Francisco, California. Slide 7, 15.
2. Mathews SJ, Stellarex in the Treatment of the SFA and Popliteal: Late- Breaking 3-Year Data, oral presentation, NCVH June 2019.
3. Lyden S, Safety And Effectiveness Of The Stellarex DCB With Low Dose Paclitaxel: 4 Year Results from the ILLUMENATE Pivotal trial, oral presentation, LINC Jan 2020, Leipzig, Germany.
4. Laird, et al. J Am Coll Cardio. 2015;66:2329-2338. In.Pact Summary of Safety and Effectiveness Data (SSED). Medtronic In.Pact Instructions for Use, M052624T001 Rev 1F.
5. Bard Lutonix Instructions for Use, BAW1387400r3.
6. Holden A. Comparing Trials Data in the Management of Calcified Arteries. Charing Cross 2018. April 24-26, 2018; London, UK.
7. Gray WA, Mortality assessment of ptx coated balloons - patient-level meta-analysis of the ILLUMENATE clinical program at 3 years, Circulation, 140:1145-1155, Oct 2019 1.
8. Lyden, S. LINC, 2019. Long-term safety data from the Stellarex DCB program Jan. 22, 2019. Leipzig, Germany.
9. Lyden S, Safety And Effectiveness Of The Stellarex DCB With Low Dose Paclitaxel: 4 Year Results from the ILLUMENATE Pivotal trial, oral presentation, LINC Jan 2020, Leipzig, Germany.
10. Diamantopoulos A, Gupta Y, Zayed H, KatsanosK. Paclitaxel-coated balloons and aneurysm formation in peripheral vessels. J VascSurg. 2015 Nov;62(5):1320-2.
11. Schmidt A et al. First experience with drug-eluting balloons in infrapoplitealarteries: restenosis rate and clinical outcome. J Am Coll Cardiol. 2011 Sep 6;58(11):1105-9.
12. LiistroF et al. Drug-eluting balloon in peripheral intervention for below the knee angioplasty evaluation (DEBATE-BTK): a randomized trial in diabetic patients with critical limb ischemia. Circulation. 2013 Aug 6;128(6):615-21.
13. Zeller T, et al. IN.PACT DEEP Trial Investigators. Drug-eluting balloon versus standard balloon angioplasty for infrapoplitealarterial revascularization in critical limb ischemia: 12-month results from the IN.PACT DEEP randomized trial. J Am Coll Cardiol. 2014 Oct 14;64(15):1568-76.
14. Venkatasubbu GD, et al. Surface modification and paclitaxel drug delivery of folic acid modified polyethylene glycol functionalized hydroxyapatite nanoparticles. Powder Technology. 2013;235:437-442.
15. Granada JF. Future directions, clinical applications and local drug delivery technologies. Presented at the Transcatheter Cardiovascular Therapeutics (TCT) 25th Annual Scientific Symposium; October 27-November 1, 2013; San Francisco, California. Slide 7, 15.
16. Stellarex: Data on file. Spectranetics Document. 2014. Spectranetics Pre-clinical Animal Study ADO097.
17. Gray B, Safety And Efficacy Of The Stellarex DCB With Low Dose Paclitaxel - 4 Year Results from the ILLUMENATE EU RCT, Viva Late Breaking Clinical Trials, 25 June 2020.
*Complex patients refers to high rates of severe calcium, diabetes and renal insufficiency. Primary patency based on Kaplan-Meier estimates.
†† No statistically significant difference in mortality
‡Data from independent CEC (clinical events committee) adjudication of all events resulting in death across all studies
Product availability is subject to country regulatory clearance. Please contact your local sales representative to check the availability in your country.
By clicking on the link, you will be leaving the official Royal Philips ("Philips") website. Any links to third-party websites that may appear on this site are provided only for your convenience and in no way represent any affiliation or endorsement of the information provided on those linked websites. Philips makes no representations or warranties of any kind with regard to any third-party websites or the information contained therein.